|
|
|
|
“지금의 통계기법으로 찾아낸 질병 유전자 후보군은 ‘빙산의 일각’에 불과합니다. 물속에 잠긴 큰 빙산까지 발굴하기 위해 효과적인 통계 알고리즘을 개발했죠.” UNIST(총장 정무영) 생명과학부의 남덕우 교수팀은 최근 질병 유전자 후보군을 ‘정확하게 많이’ 찾아내는 통계 알고리즘(GSA-SNP2)을 개발했다. 이 알고리즘은 만 명 이하의 적은 유전체 데이터만 있어도 효과적으로 작동한다. 질병 유전자 후보군을 발굴하는 비용과 시간을 크게 줄일 방법으로 주목받고 있다. 남덕우 교수는 “몇 천 명 단위에서도 의미 있는 유전자 그룹을 찾아낼 수 있는 저비용 고효율 통계분석도구”라며 “이 알고리즘을 통해 신약 개발을 위한 유전자 표적을 발굴하거나 질병에 대한 이해를 더 빠르게 진행시킬 수 있다”고 강조했다. 사람의 DNA 염기서열은 조금씩 다르게 나타나는데, 이로 인해 질병에 대한 감수성 등 다양한 표현형이 결정된다. 이런 염기서열의 차이를 ‘스닙(SNP)’이라고 하며, 대규모 유전체 데이터를 통계적으로 분석하면 특정 질병과 관련된 스닙(SNP)들을 찾을 수 있다. 천문학적인 비용과 시간을 들여서 데이터를 생산해도, 현재 사용하는 통계분석 방법들은 유의미한 스닙(SNP)을 많이 찾지 못한다. 질병 유전자가 아닌데, 질병 유전자로 판단하는 ‘허위양성’ 결과를 엄격하게 통제하도록 설계됐기 때문이다. 결국 수만 명의 유전형 데이터를 생산하고, 수십만에서 백만 개 이상의 스닙(SNP)을 대상으로 분석해도, 질병 유전자 후보군 수십 개 정도를 얻는 데 그치게 된다. 남 교수는 “허위양성을 통제해서 정확한 결과를 얻는 것도 중요하지만, 너무 많은 스닙(SNP)을 걸러내면 실제 신약개발 등에서 효용성이 낮아진다”며 “질병 유전자 후보군을 많이 발굴해낼 수 있는 ‘통계적 예측력(statistical power)’도 높여야 실용적인 통계 알고리즘이 된다”고 말했다. |
남 교수팀은 허위양성을 잘 통제해 정확한 결과를 얻으면서도 통계적 예측력을 높이는 알고리즘 개발을 목표로 삼았다. 이를 위해 ‘유전자 그룹(pathway) 상관관계 분석법’을 활용하면서 유전자 스코어에 큐빅 스플라인(cubic spline) 이라는 수학적 보정을 적용했다. 유전자 그룹은 특정 기능을 수행하는 데 관여하는 유전자 집단이다. 이들은 수백에서 수천가지 그룹들로 선별돼 데이터베이스로 정리돼 있다. 이 정보를 이용하면 개별 스닙(SNP) 비교에서는 놓쳤던 의미를 새롭게 찾을 수 있다. 남 교수팀은 이 기법을 쓰면서, 이미 질병과 상관관계가 높게 나타난 스닙(SNP)들은 제외하고 유전자 스코어를 보정함으로써 통계적 예측력을 높였다. 남 교수는 “질병 유전자 후보로 강하게 판단되는 스닙(SNP)을 빼면, 임의의 유전자 분포(영 분포)를 얻게 된다”며 “이 상태에서 다시 통계적으로 유의미한 걸 찾아내도록 설계했기 때문에 기존 방법들보다 2배 이상 예측력이 높아졌다”고 설명했다. 그는 이어 “새로운 통계 알고리즘을 적용하면 다양한 질병 유전자 그룹들을 다수 발굴할 수 있을 것”이라며 “신약개발 또는 스닙(SNP)분석 관련 연구기관이나 기업에서 활용하면 질병 치료에 기여하는 유용한 도구가 될 것”이라고 덧붙였다. 이번 연구는 UNIST 생명과학부의 윤소라 대학원생과 응우옌 하이(Nguyen C. T. Hai) 박사가 공동 1저자로 수행했으며, 포스트게놈 다부처유전체사업에서 연구비를 지원 받았다. 연구결과는 영국 옥스퍼드대학 출판사에서 발행하는 저명한 생물학 저널 ‘뉴클레익 에시드 리서치(Nucleic Acids Research, IF: 10.162)’ 3월 19일자 온라인판에 게재됐다. (끝)
|
|
|
|
[붙임] 연구결과 개요 |
1. 연구배경DNA를 이루는 염기는 아데닌(A), 시토신(C), 구아닌(G), 티민(T) 네 가지다. 이들 염기는 일정한 순서대로 배열되며, 이를 염기서열이라고 한다. 사람은 30억 염기쌍으로 된 유전체를 가지는데, 이중 하나의 염기서열이 사람마다 조금씩 다르게 나타날 수 있다. 이런 현상을 ‘단일염기다형성(Single Nucleotide polymorphism)’이라 부르며, 스닙(SNP)으로 읽는다. 많은 경우, 스닙들이 특정 질병에 대한 취약성이나 키, 지능 등 개인 간 표현형의 차이를 결정하고 있다. 유명한 예로서, 배우 안젤리나 졸리가 BRCA 유전자에 높은 위험성을 가진 변이가 발견되어 예방을 위해 유방절제술을 받은 바가 있다. 특정 질병과 상관관계가 높은 스닙(SNP)들을 발굴하려면 보통 수십만 개에서 백만 개 이상의 스닙(SNP)들에 대해서 수만 명의 유전형 데이터를 생산해 분석하는 데 이를 ‘전장유전체상관분석(GWAS)’이라고 한다. 이런 방식은 데이터 생산에 천문학적인 비용이 들어가고, 많은 시간이 소요된다. 그럼에도 불구하고, 부정확한 정보를 엄격하게 걸러내는 통계분석을 수행하고 나면 통계적으로 유의미한 스닙(SNP)는 보통 수십 개 정도에 불과하다. 이런 결과로는 질병의 유전적 요인을 아주 일부만 설명할 수 있다. 이런 상황은 ‘빙산의 일각’으로 비유되는데, 수십 개의 유의미한 스닙(SNP)만 물 밖으로 드러나서 통계적으로 ‘발견’할 수 있다는 의미다. 실제로 유전적 요인 대부분은 수면 아래에 잠겨있는 상황이라고 할 수 있다. 전통적 통계분석 방법의 낮은 통계적 예측력(statistical power)을 극복하기 위해 10여 년 전부터 ‘유전자 그룹(pathway)’ 단위의 상관관계 분석이 도입됐다. 유전자 그룹은 특정 세포기능을 수행하는 데 관여하는 유전자들의 집단을 말하는데, 수백에서 수천가지의 기능 그룹들이 선별돼 데이터베이스에 분류돼 있다. 이 방법은 개별 스닙(SNP)의 상관관계는 비교적 약하게 나타나더라도 유전자 그룹으로 묶어서 분석하면 통계적으로 유의한 상관관계를 새롭게 찾을 수 있다는 점에 착안하고 있다. 대표적인 통계 알고리즘은 ‘마젠타(MAGENTA)’와 ‘마그마(MAGMA)’가 있다. 마젠타의 경우, 질병과 관련 없는 유전자를 관련 있다고 표현하는 ‘허위양성’ 예측을 엄격히 통제하는 특징이 있다. 하지만 질병과 관련 있을 것으로 예측되는 유전자 후보군을 발굴하는 ‘통계적 예측력’이 낮아 유의한 유전자 그룹을 발굴하지 못하는 경우가 많다. 마그마는 비교적 최근에 개발된 방법으로서 허위양성 예측을 엄격히 통제하면서도 현재까지 가장 높은 수준의 통계적 예측력을 보여주고 있다. |
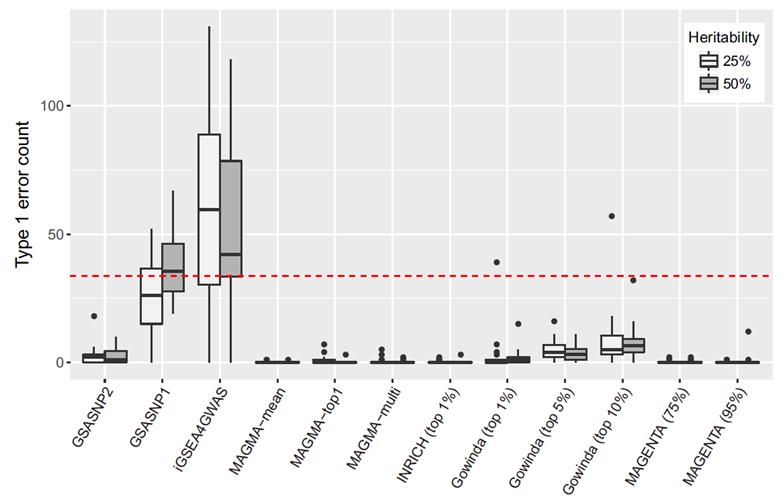
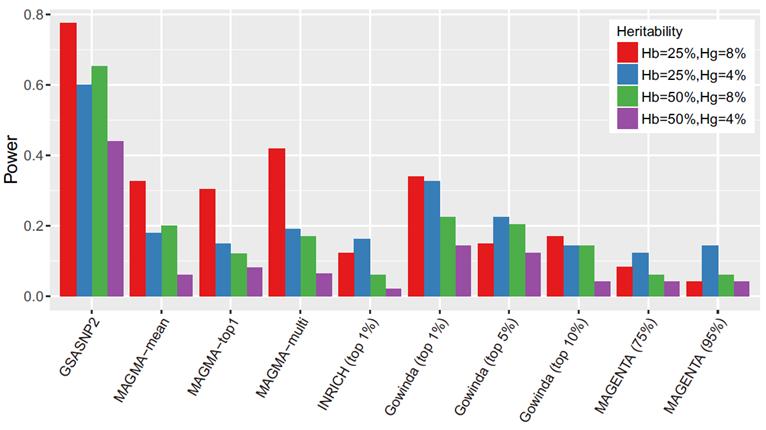
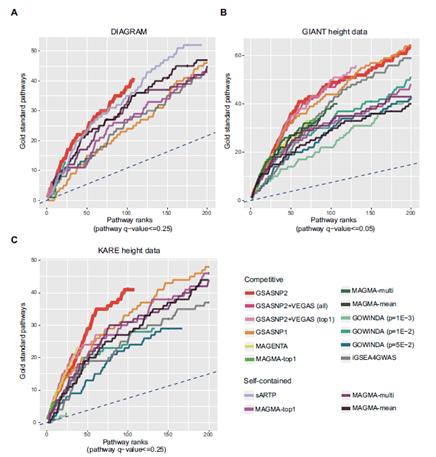
2. 연구내용이번 연구에서는 유전자 그룹 분석법을 획기적으로 발전시킨 통계적 알고리즘(GSA-SNP2)을 개발했다. 이 알고리즘은 기존 마젠타에 버금갈 정도로 허위양성예측을 통제하면서 최고 수준의 예측력을 보였다. (그림1, 그림2 참고) GSA-SNP2에서는 각각의 스닙(SNP)들을 가장 가까운 유전자에 배정한 후 유전자 스코어를 구하는데, 배정된 스닙(SNP)이 많을수록 유전자 스코어가 증가하는 경향을 보이게 된다. 이러한 패턴을 단조증가 큐빅 스플라인 (cubic spline)이라는 수학적 알고리즘으로 보정함으로써 허위양성예측을 대부분 억제할 수 있었다. 큐빅 스플라인은 그래프에서 주어진 점을 매끄럽게 연결하는 알고리즘인데, 여기서는 단조증가 함수를 사용해 상승하는 경향만 나타내도록 했다. 이 알고리즘의 강한 예측력 또한 이러한 수학적 보정의 결과이다. 특히, 높은 질병연관성 스코어를 갖는 유전자들을 사전에 배제한 후, 유전자 스코어를 보정한 것이 이 알고리즘의 주요한 특징이다. 통계적으로 유의미한 값들을 제외하고, 임의의 값들 (영 분포)만으로 보정하기 때문에 통계적 예측력이 높아지는 것이다. 1000 Genomes에서 제공하는 실제 유전체 데이터와 선형모델을 이용한 대규모 시뮬레이션 실험에서 GSA-SNP2는 거의 모든 허위양성 예측을 억제하면서도 기존의 방법들보다 2배 이상의 높은 예측력을 나타냄을 확인할 수 있었다. 이러한 예측력은 ‘2형 당뇨’와 ‘성인 신장(키)’등 실제 데이터 분석에서도 확인할 수 있었는데, 문헌에서 찾은 검증된 유전자 그룹들을 기준으로 각 알고리즘들의 예측성능을 비교한 결과, GSA-SNP2는 마그마나 마젠타 등 기존의 알고리즘들보다 뛰어난 통계적 예측력을 보임을 알 수 있었다. (그림3) |
3. 기대효과GAS-SNP2는 적은 비용과 시간으로 다양한 질병 관련 유전자 그룹들을 다수 발굴해 낼 수 있다. 기존의 알고리즘들이 수천 정도의 비교적 적은 샘플의 데이터를 사용했을 경우 예측력이 현저히 떨어지는 것에 비해, GSA-SNP2는 적은 샘플의 데이터일수록 상대적으로 월등한 예측력을 보인다. 이를 통해 중소규모의 유전체 연구에서, 비용을 크게 줄이면서도 실질적인 결과물을 얻을 수 있을 것으로 기대된다. 또한, 기존 통계 알고리즘들이 수십 분에서 수주까지 계산시간을 소요하는 데에 비해 분석시간도 대폭 짧아져서 2분 이내에 결과를 얻을 수 있어서, 질병연구와 신약개발의 시간을 절약해 줄 수 있다. |
|
[붙임] 논문저자 1문 1답 |
질문1. 생명과학 연구에 수학과 통계를 적용하면 어떤 이득이 있을지?유전체처럼 대규모 데이터를 얻었을 때, 그것을 활용할 수 있는 과학적인 근거는 ‘통계’뿐입니다. 대규모의 데이터에서 일정한 경향성을 찾아내고, 그것을 토대로 실험을 진행해 검증하면서 생명현상을 이해할 수 있어요. 통계적인 방법으로 생명의 신비를 이해하는 지름길을 열어준다고 생각해도 좋을 것 같습니다. 예를 들어, 자연은 어떤 유전자가 질병 관련 유전자인지 알고 있습니다. 하지만 사람들이 그것을 일일이 실험하면서 찾아내기는 너무 막막한 일입니다. 이때 수많은 사람들의 DNA 염기서열을 비교하면 공통점과 차이점을 찾아낼 수 있어요. 어떤 질병을 가진 사람들에게만 특정 위치에서 특정 염기서열이 다르게 나타났다면 그곳이 질병과 관련되었을 가능성이 높지요. 질문2. 국내에 생물통계학이나 생물정보학 분야를 연구하는 사람이 많은지?외국에는 생물통계학이나 생물정보학 분야의 연구가 훨씬 활발합니다. 유전체 빅데이터 분석 프로젝트에 꼭 필요한 분야들이니까요. 특히 대형 제약사에서는 신약 개발을 위한 표적 유전자를 찾을 때 통계 알고리즘이 중요하게 쓰여서 이 분야 연구자들이 귀한 대접을 받고 있습니다. 아쉽게도 국내에는 제약산업 규모가 작아서 상대적으로 이 분야 연구자가 적은 편입니다. 그래도 최근에는 정부와 의대 쪽에서 유전체 연구에 투자를 늘리는 추세라 생물정보학 연구도 점차 늘어나고 있습니다. 그래도 아직은 해외에서 개발해둔 알고리즘과 프로그램을 도입해서 분석하는 경우가 많습니다. 저처럼 새로운 통계 알고리즘을 개발하는 연구자는 많지 않습니다. 질문3. 수학을 전공하고 생물학 분야로 뛰어든 이유는 무엇인지?수학, 통계로 세상을 이롭게 하는 일을 하고 싶었습니다. 카이스트를 졸업하던 즈음에 한국생명공학연구원에 생물정보학을 연구하는 그룹이 처음 꾸려졌습니다. 거기서 응용수학 전공자를 뽑는다고 해서 들어갔고, 생물정보학을 시작했습니다. 생물은 물론 컴퓨터 프로그램에도 서툴러서 처음에는 애를 좀 먹었죠. 그런데 연구를 하면 할수록 적성에 맞는다는 생각이 듭니다. 여러 지식을 종합해서 유용한 결론을 내는 것이 아주 재미있고 보람도 있습니다. 앞으로도 유전체 연구에 중요한 영향을 주는 멋진 통계 알고리즘을 선보일 계획입니다. |
|
[붙임] 용어설명 |
1. 뉴클레익 에시드 리서치(Nucleic Acids Research, NAR)영국 옥스퍼드대학 출판사에서 출간하는 생물학 학술지(journal)로, 핵산(nucleic acids) 연구 관련해서 저명한 위치에 있다. 물리학과 화학, 생화학, 생물학, 계산 및 통계적 접근방법들을 폭넓게 다루고 있다. 2016-2017 영향력 지수(Impact Factor)는 10.162이다. 2. 단일염기다형성(Single Nucleotide polymorphism, SNP)DNA를 이루는 염기는 4가지(A, T, G, C)가 있으며, 이들이 배열된 순서(염기서열)이 다른 하나가 나타날 때 유전적 변화 또는 변이를 ‘단일 핵산염기 다형현상(Single Nucleotide polymorphism)’이라고 한다. 약자로 SNP로 쓰며, 스닙이라고 읽는다. 사람의 유전체는 30억 염기쌍으로 이뤄진다. 이 중 1% 이상의 빈도로 존재하는 2개 이상의 대립 염기서열이 발생하는 위치를 SNP라 하며, 대립유전자형이 5% 이상 빈도로 존재하면 ‘평범한 다형성(common polymorphism)’이라고 하며, 1~5%인 경우 ‘희소한 다형성(rare polymorphism)’으로 분류한다. 3. 허위양성(false positive)‘1종 오류(type I error)’라고도 하며, 실제로는 ‘음성’인데 검사 결과는 ‘양성’이라고 나오는 경우를 이르는 통계학적 용어다. 예를 들어, 질병 관련 유전자를 판별하는 통계적 방법을 적용했을 때, 질병 유전자가 아닌데 질병 유전자로 예측을 할 때 사용하는 용어다. 반대로 ‘허위음성(false negative, 2종 오류)는 질병 유전자가 맞는데 질병 유전자가 아니라고 판별하는 경우에 해당된다. 허위양성과 허위음성은 유의 수준에 따라 일반적으로 반비례하는 경향이 생기며 이 두 가지 오류를 최소로 줄이는 통계적 방법의 개발이 중요하다. 4. 통계적 예측력(Statistical power)허위음성이 적을 때, 즉 대부분 실제 질병 유전자들에 대해서 질병 유전자가 맞다고 예측할 경우 통계적 예측력이 높다고 한다. |
|
[붙임] 그림 설명 |
|
그림1. 허위양성통제 시뮬레이션 결과 비교: GSA-SNP2(왼쪽 끝)가 마그마(왼쪽 3~6번째), 마젠타(오른쪽 끝) 등과 비등한 수준의 허위양성통제를 보여주고 있다.
그림 2. 통계적 예측력 시뮬레이션 비교: GSA-SNP2(왼쪽 끝)가 기존 방법들에 비해 2~5배의 높은 통계적 예측력을 보여주고 있다.
그림3. 2형 당뇨와 성인 신장(키) 데이터 분석에서 GSA-SNP2(빨간색 그래프)가 문헌에 검증된 유전자 그룹들을 다른 방법들보다 더 잘 찾아줌을 나타내고 있다. |
|
|
UNIST 홍보팀 news@unist.ac.kr TEL : 052)217-1230FAX : 052)217-1229 |