|
|
|
|
|
똑같은 암을 앓는 환자라도 항암제 효능은 달라질 수 있다. 개인별 유전변이 등에 따라 항암제의 반응성이 다르게 나타나기 때문이다. 이런 환자별 결과를 미리 알려주는 인공지능(AI) 기술이 개발돼 주목받고 있다. UNIST(총장 이용훈) 바이오메디컬공학과 이세민 교수팀은 고려대 정원기 교수팀, 한양대 서지원 교수팀과 공동으로 ‘다중 오믹스 데이터 기반의 환자 맞춤형 항암제 반응성 예측을 위한 기계학습 모델’을 개발했다. 연구진은 대규모 항암제 반응성 데이터와 다중 오믹스 데이터를 활용해 기존의 항암제 반응성 예측 모델보다 훨씬 우수한 성능을 얻었다. 네트워크 임베딩 기술과 최신 딥러닝 모델이 적용된 덕분이다. 암은 대표적인 유전체(Genome) 관련 질병, 즉 ‘게놈 병’이다. 사람마다 가진 ‘생명의 설계도’인 유전체에 계속 변이가 축적되면서 질병이 발생한다는 뜻이다. 암 조직에서는 유전자 발현 양상도 정상조직과는 달라진다. 이러한 유전변이와 유전자 발현 프로파일(Profile)은 동일 암종의 환자 간에도 상당한 차이를 보이는데, 이는 환자 특이적 항암제 반응성과 유의미한 연관성이 있다고 알려져 있다. 이에 따라, 최근에는 암 환자 특이적 유전변이나 유전자 발현 양상 등을 아우르는 다중 오믹스 데이터를 기반으로 ‘환자 맞춤형 항암제 반응성 예측 모델’을 개발하는 시도가 많다. 그러나 이런 모델 학습을 위한 생물학 데이터는 종류와 인자가 많은 데 비해 샘플 수는 부족해 기계학습 모델의 정확도를 높이는 데 한계가 있다. 연구진은 이를 극복하기 위해 ‘네트워크 임베딩 기술’을 적용해 다차원 데이터 간의 상관관계를 효과적으로 반영했다. 먼저 암세포에서 파생된 세포주와 항암제, 유전자를 ‘노드(node, 연결점)’로 삼았다. 다음으로 각 노드를 연결해 엣지(edge, 연결선)를 만들었다. 엣지를 통해서는 항암제 반응성(세포주-항암제)이나 유전자 변이(세포주-유전자), 단백질 상호작용(유전자-유전자)에 대한 정보를 얻을 수 있다. 노드와 엣지로 형성된 네트워크 세트의 상관관계를 반영한 ‘임베딩 벡터’의 추출이 이번 연구의 핵심이다. 임베딩 벡터를 이용하면 각 노드의 대푯값을 알 수 있어 고차원적인 데이터도 효과적으로 다룰 수 있기 때문이다. 연구진은 임베딩 벡터를 AI 기법인 심층신경망으로 학습시켜 환자맞춤형으로 항암제 효능을 도출했다. 제1저자인 고려대 이강근 박사는 “저항성 위주로 편향된 반응성 데이터를 보완하기 위한 다양한 인공지능 기법을 적용했다”며 “새 모델의 항암제 반응성 예측 성능은 기존 모델보다 크게 향상된 93% 정도의 정확도로 나타났다”고 설명했다. 공동 제1저자인 한양대 조동빈 연구원은 “고차원 다중 오믹스 데이터에 존재하는 요소끼리 상호작용을 효과적으로 추출하는 네트워크 임베딩 기술을 비롯한 심층신경망 등을 통해 우수한 성능을 달성했다”고 전했다. 또 다른 공동 제1저자인 UNIST 장진호 박사는 “이 기술은 암 환자에게 적합한 약물의 후보를 제안함으로써 맞춤 치료 가속화할 것”이라고 기대했다. 이번 연구결과는 생명정보학 분야 최고 학술지인 ‘브리핑스 인 바이오인포메틱스(Briefings in Bioinformatics)’에 공개됐으며, 한국연구재단의 ‘차세대정보컴퓨팅기술개발사업’과 ‘대학중점연구소지원사업’에서 지원했다. (논문명: RAMP: response-aware multi-task learning with contrastive regularization for cancer drug response prediction) |
|
|
|
[붙임] 연구결과 개요 |
1. 연구배경정밀의학의 목표는 유전체 데이터와 임상 정보를 통합해 환자의 치료 요법을 최적화하는 것이다. 근래 정밀의학은 정밀종양학의 발전 덕분에 암 환자의 예후를 개선할 수 있는 새로운 치료법들을 제시하고 있다. 그러나 소수의 환자에게만 임상시험과 치료법의 혜택이 제공되고 있다. 그 이유 중 하나는 각 환자의 유전체 데이터를 표현형으로 번역하는 것이 어렵기 때문이다. 더욱이 유전체 데이터의 생산은 비용이 많이 들고 각 환자에게 투여할 수 있는 약물도 제한된다. 이에 따라 종양 샘플에서 파생된 세포주를 활용한 항암 약물 스크리닝을 활용하는 방향이 추진되고 있다. 이러한 흐름에 힘입어 고속 약물 스크리닝 기술이 개발됐으며 암 세포주에서 다수의 항암제 스크리닝이 가능해졌다. 또한, 다양한 암 세포주 및 항암제를 활용한 대규모 암 유전체 프로젝트들이 추진됐으며 ‘암 약물 민감성 관련 유전체학(Genomics of Drug Sensitivity in Cancer, GDSC)’과 ‘암 세포주 백과사전(Cancer Cell Line Encyclopedia, CCLE)’와 같은 공개 데이터베이스들이 형성됐다. 이러한 데이터베이스는 풍부한 항암 약물 반응성 정보와 유전체, 전사체 및 후성 유전체 데이터를 포함한 다중오믹스 데이터를 제공함으로써 약물 반응 예측 모델의 개발에 박차를 가했다. 데이터베이스를 통해 대규모 데이터들이 공유되면서 이를 활용해 약물 반응성을 예측하는 기계학습 모델의 개발이 활발하게 일어나고 있다. 특히 세포주의 유전체 정보와 약물 반응성 간의 상관관계를 이해하기 위해 세포주의 유전체 정보, 약물 반응성 정보, 그리고 단백질 간의 상호작용 정보를 포함하는 대규모 네트워크를 기반으로 다양한 예측 모델들이 개발되고 있다. 하지만 근래 연구들은 다양한 세포주 유전체 정보 및 약물 유사성 정보와 같은 폭넓은 데이터의 활용에 한계가 있다. 이러한 기존 연구의 한계점을 보완하기 위해 추가적인 임베딩 및 딥러닝 기법을 토대로 새로운 약물 예측 모델인 ‘암 약물 반응 예측을 위한 대조 정규화를 사용한 응답 인식 멀티태스크 학습(Response-Aware Multi-task Learning with Contrastive Regularization for Cancer Drug Response Prediction, RAMP)’를 개발했다. |
2. 연구내용이번 연구에서는 GDSC 세포주의 ‘유전체 정보’와 ‘약물 반응성 정보’ 및 iRefIndex 데이터베이스의 ‘단백질 간 상호작용 정보’를 활용해 대규모 네트워크를 구축했다. 해당 네트워크의 각 노드(node, 연결지점)에 대해, 더욱 정확한 임베딩 벡터를 추출하기 위해 약물 저항성 정보를 우선 활용하는 ‘응답 인식 네거티브 샘플링(response-aware negative sampling, RA-NS)’을 적용했다. 또한, 추출한 임베딩 벡터가 실제 세포주에 대한 약물의 반응성을 얼마나 잘 반영하는지 평가하기 위해 ‘임베딩 점수’라는 지표를 고안했다. 추가로 다중 작업 학습에 적합한 ‘약한 지도에 따른 대조 정규화(soft-supervised contrastive regularization, SSCR)’을 활용해 다중 약물 예측 모델을 개발했다. 오버샘플링 또는 적응적 가중치 손실 없이 심층 신경망(Deep Neural Network, DNN)의 멀티태스킹 아키텍처를 통해 저항성 정보가 주를 이루는 약물 반응성 정보의 불균형 분류를 성공적으로 완화했다. 이렇게 새로운 구성 요소(RA-NS, SSCR, Monte Carlo dropout)를 토대로 형성한 멀티태스킹 아키텍처가 교차 검증을 통해 다른 기존 기계학습 기술보다 개선된 성능을 보이는 것을 확인했다. |
3. 기대효과이번 연구에서는 약물 반응성 네트워크 분석 결과를 이용해 약물 반응성을 정확하게 예측하는 방법을 개발했다. 첫째, 약물 반응성 네트워크에 대해 더욱 정확한 임베딩 벡터를 식별하기 위해 RA-NS를 도입했으며, RA-NS를 평가하기 위해 임베딩 벡터가 실제 약물 반응성을 얼마나 정확하게 반영하는지를 측정하는 임베딩 점수를 정의했다. 해당 지표를 활용해 평가한 결과, RA-NS로 추출한 임베딩 벡터가 테스트에 활용된 전체 약물의 76.23%에 대해 기존의 네거티브 샘플링(negative sampling, 필요한 일부만 우선 활용하는 기법)으로 추출한 임베딩 벡터보다 더 높은 임베딩 점수를 가지는 것을 확인할 수 있었다. 또한, DNN 모델의 약물 반응 예측 성능을 높이기 위해 멀티태스킹 학습에 적합한 새로운 SSCR을 적용했으며, 다른 기존의 멀티태스킹 학습 방식과 달리 오버샘플링 없이 불균형 분류 문제를 극복할 수 있었다. RAMP는 GDSC 데이터 기반의 교차 검증을 통해 93%의 정확도를 보였다. 또한, GDSC 데이터베이스에 누락돼 있는 세포주와 약물 간의 반응성까지 정확히 예측하는 것을 기존 반응성 연구와의 비교를 통해 확인했다. 최종적으로 RAMP는 실제 암 환자에게 적합한 약물의 후보를 제안하는 데 유용할 것으로 보인다. |
|
[붙임] 그림설명 |
|
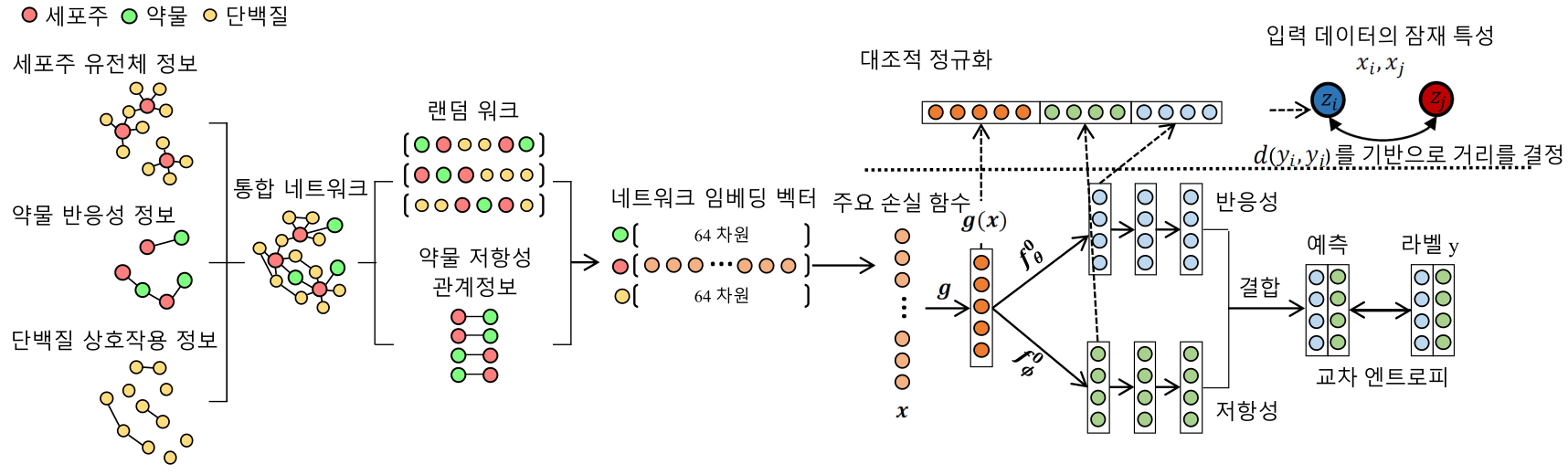
그림1. 환자 맞춤형 항암제 반응성 예측 모델 구축의 작업 흐름도RAMP의 형성에 대한 구조를 보여주는 그림은 크게 두 가지의 주요 단계로 나누어진다. 먼저 세포주의 유전체 정보, 약물 반응성 정보, 그리고 단백질 간의 상호작용 정보를 포함하는 대규모 네트워크에 대해 RA-NS를 기반으로 각 노드에 대한 임베딩 벡터를 학습하는 과정이 진행된다. 두 번째로, 임베딩 벡터로부터 ‘대조적 정규화(soft-supervised contrastive regularization)을 활용해 심층신경망의 멀티태스킹 아키텍처를 학습시킨다. |
|
|
UNIST 홍보팀 news@unist.ac.kr TEL : 052)217-1230FAX : 052)217-1229 |