|
|
|
|
|
염색질 면역 침전(Chromatin immunoprecipitation, ChIP)은 특정 단백질의 결합 위치를 조사하는데 널리 사용된다. 특히 엑소뉴클리아제(exonuclease)를 활용한 최신 실험 기술인 ChIP-exo을 통해 고해상도로 결합 부위를 식별할 수 있다. 하지만 목표로 하는 단백질이 실제로 DNA에 결합하는 부위인 피크에 대한 판별 작업은 연구자의 노동집약적 추가 확인 단계가 필수적이다. 이는 대용량의 데이터를 신속·정확하게 처리하지 못하는 한계가 있었다. UNIST(총장 이용훈) 에너지화학공학과 김동혁 교수팀은 이런 문제점을 해결하기 위해 딥러닝 기반의 ChIP-exo 피크 선별 소프트웨어인 DEep-learning Optimized ChIP-exo peak calling SUite(DEOCSU)를 개발했다. DEOCSU는 참조 서열에 정렬된 ChIP-exo 데이터를 통해 피크 후보를 먼저 감지한다. 감지된 각각의 신호를 이미지 데이터로 변환한 후 학습된 데이터를 통해 이미지를 작은 단위로 쪼개어 각 부분을 분석하는 기법인 컨볼루션 신경망을 사용하여 실제 피크를 선별한다. 선별된 각 피크는 위치 최적화와 결합 크기 등을 추정할 수 있다. 해당 결과 데이터는 자체 개발 시각화 소프트웨어인 MetaScope를 통해 확인한다. 대장균 K-12 MG1655 균주의 ChIP-exo 데이터를 기반으로 학습된 DEOCSU의 모델은 학습에 사용된 데이터 뿐만 아니라 미지의 ChIP-exo 데이터에 대해서도 정확하게 피크를 선별해줬다. 공개 데이터베이스(EcoCyc와 proChIPdb)의 정보와 선행된 연구 사례를 이용하여 기존에 공개된 소프트웨어(ChExMix, MACE, MACE-elite, PeakXus)와 비교했을 때도 우수한 성능을 보였다. 특히, 원핵생물 유래의 ChIP-exo 데이터 뿐 아니라 진핵생물과 고세균에서의 분석에서도 그 성능이 유지가 되어 그 범용성 역시 확인됐다. 김동혁 에너지화학공학과 교수는 “단백질과 DNA의 상호 작용을 고해상도로 식별할 수 있는 유용한 이점에도 불구하고 분석의 어려움으로 인해 ChIP-exo 실험 기술의 사용이 제한되었었다”며 “이번 DEOCSU의 개발로 분석에 대한 연구자의 부담감을 극복시킴으로써 관련 연구의 진행 속도를 가속화할 수 있을 것이다”고 전했다. 이번 연구는 과학기술정보통신부의 바이오·의료기술개발사업과 동그라미 재단의 혁신 과학기술 센터 및 프로그램 공모사업의 지원으로 수행됐다. 연구 성과는 생물정보학 연구 권위지인 브리핑스 인 바이오인포메틱스 (Briefings in Bioinformatics)에 1월 25일자로 출판됐다. (논문명: Deep-learning optimized DEOCSU suite provides an iterable pipeline for accurate ChIP-exo peak calling.)
|
|
|
|
[붙임] 연구결과 개요 |
1. 연구배경염색질 면역 침전(ChIP)은 DNA와 결합 단백질(예: 전사 인자(TF))의 결합 위치를 유전체 수준에서 조사하는 데 널리 사용되어 왔다. ChIP-exo는 신호 대 잡음비를 증가시키고 연구자들이 단일 염기 쌍 수준의 고해상도 결합 부위를 식별할 수 있게 하지만, 실제 피크를 선별하기 위한 피크 호출 단계는 ChIP-exo 데이터 분석의 주요 속도 제한 단계이며 시간이 많이 걸리고 노동 집약적이다. ChIP-exo 데이터를 분석하여 해당 단백질의 결합 위치를 선별하기 위한 다양한 계산 도구가 개발되었다. 그럼에도 불구하고, 이들은 단순히 몇 개의 데이터 세트를 사용하여 검증되었고, 너무 많은 오탐지 결과를 제공하므로 추가적인 수동 선별 단계가 불가피하다. |
2. 연구배경본 연구에서는 컨볼루션 신경망(CNN)을 사용하여 ChIP-exo 데이터의 실제 피크를 선별하는 DEEP-learning Optimized ChIP-exo Peak Calling Suite(DEOCSU)를 개발하였다. DEOCSU는 대장균(E. coli) str. K-12 MG1655 균주에서 시그마 인자(RpoD, RpoH, RpoN, RpoS) ChIP-exo 데이터의 이미지 변환 피크를 사용하여 훈련되었다. DEOCSU의 최대 호출 성능은 6개 대장균주(BL21(DE3), CFT073, Crooks, O157:H7 EDL933, HS, K-12 W3110) 및 기타 박테리아(Klebsiella, Shigella)의 RpoN ChIP-exo 데이터 세트를 사용하여 MACE, MACE-elite, ChExMix 및 PeakXus를 포함한 공개된 소프트웨어와 비교되었다. 이후 공개 데이터베이스인 proChIPdb에서 얻은 ChIP-exo 데이터와 해당 전사 인자의 선별된 ChIP-exo 피크를 사용하여 성능을 검증했다. 추가적으로, 진핵생물과 고세균에 대한 DEOCSU 적용의 확장성은 NCBI 또는 EML-EBI의 ChIP-exo 데이터 세트를 사용하여 입증되었다. 결과적으로 DEOCSU는 높은 정확도, 민감도, 낮은 음의 선별값으로 서로 다른 유기체의 데이터에서도 명확하게 ChIP-exo peaks를 선별하는 것으로 나타났다. 정확한 피크 호출 성능 외에도 DEOCSU에는 몇 가지 추가적인 이점이 있다. 첫째, DEOCSU는 임계값을 조정할 수 있도록 실제 피크 신뢰도에 대한 확률 값을 제공한다. 따라서 이 사용자 정의 가능 기능을 통해 연구자는 ChIP-exo 분석 목적에 따라 유연하게 대처 할 수 있다. 둘째, 미래에 생성될 새로운 데이터의 추가적인 훈련을 통해 보다 다양한 결합 패턴을 탐지하도록 성능을 최적화할 수 있다. 마지막으로 생물학적 복제 데이터가 없더라도 단일 ChIP-exo 데이터의 분석을 통해 피크 호출을 수행할 수 있다. 결과적으로, DEOCSU는 심층 컨볼루션 신경망 접근법을 사용하여 실제 피크의 이미지 패턴을 구별함으로써 ChIP-exo 데이터 세트에서 DNA 결합 부위에 대한 매우 정확한 선별을 제공할 수 있다. |
3. 기대효과유전체 수준에서 특정 단백질의 결합 위치를 고해상도로 식별할 수 있는 유용한 이점에도 불구하고, ChIP-exo는 시간이 많이 걸리고 노동 집약적인 피크 선별 단계를 필요로 하므로 사용이 제한된다. 여러 피크 호출 도구의 개발에도 불구하고 잘못된 피크 선별으로 인해 모티프 검색이나 추가 선별 과정이 실패하는 문제는 여전히 남아 있다. DEOCSU는 이러한 ChIP-exo 분석의 어려움을 완화함으로써 관련 연구의 진행속도를 가속화할 수 있을 것으로 기대된다. |
|
[붙임] 용어설명 |
1. 내염색질 면역 침전(Chromatin immunoprecipitation, ChIP)DNA-단백질의 결합 상태를 보기 위한 실험 방법으로, 특정 단백질에 의해 회수된 DNA 조각들의 위치를 파악함으로써 해당 단백질이 유전체의 어느 부위에 결합하였는지를 확인할 수 있음. |
2. 엑소뉴클리아제(exonuclease)핵산의 맨 끄트머리부터 뉴클레오타이드(포스포다이에스터의 3’또는 5’결합)를 한 번에 하나 씩 끊는 효소. |
|
[붙임] 그림설명 |
|
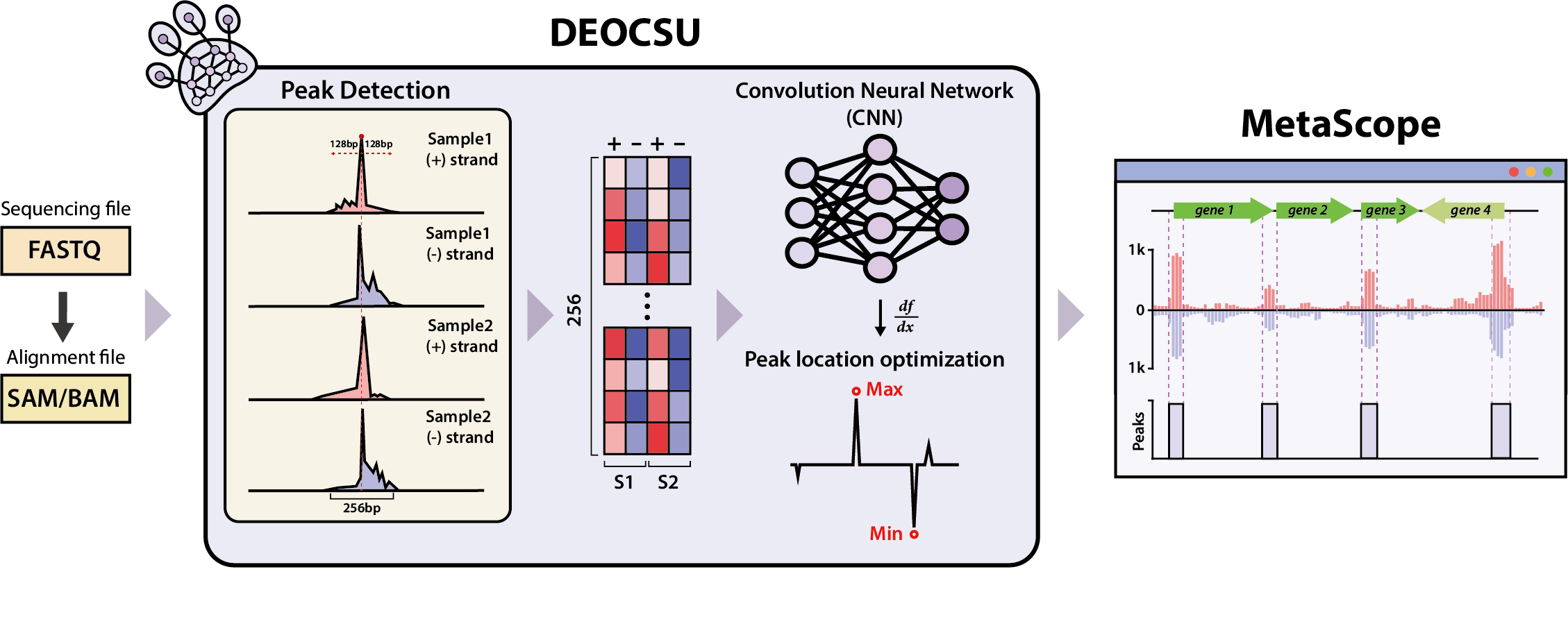
그림 1. DEOCSU의 개요.DEOCSU의 전체적인 워크플로우는 피크 후보의 감지, 각 신호에서 이미지 데이터로의 변환, 학습된 컨볼루션 신경망을 사용한 실제 피크의 선별, 피크 위치 최적화 및 결합 크기 추정의 주요 단계로 이루어지며 이후 결과데이터는 MetaScope를 사용하여 유전체 전체에서의 분포를 시각화할 수 있음. |
|
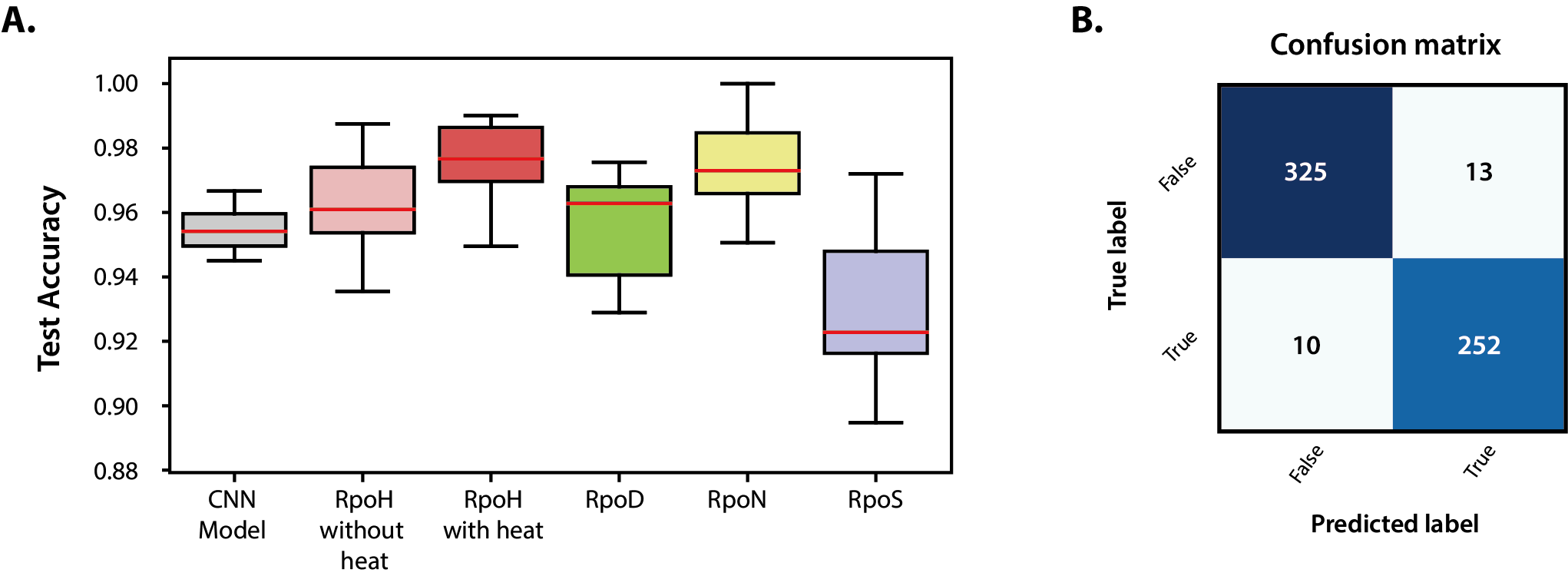
그림 2. 대장균 K-12 MG1655 시그마 인자의 ChIP-exo 데이터를 이용한 DEOCSU의 성능 검증.(A) DEOCSU에서 사전 훈련된 모델의 테스트 정확도와 각 시그마 인자 데이터의 테스트 정확도. DEOCSU의 모델이 높은 정확도(96.2%), 정밀도(95.1%) 및 재현율(96.2%)을 가지고 있음을 알 수 있으며, 10회 반복으로 무작위 재표본 추출을 통한 성능 분석을 진행하였을 때 정확도의 평균이 92.3% 이상으로 나타난 것을 확인할 수 있음. |
|
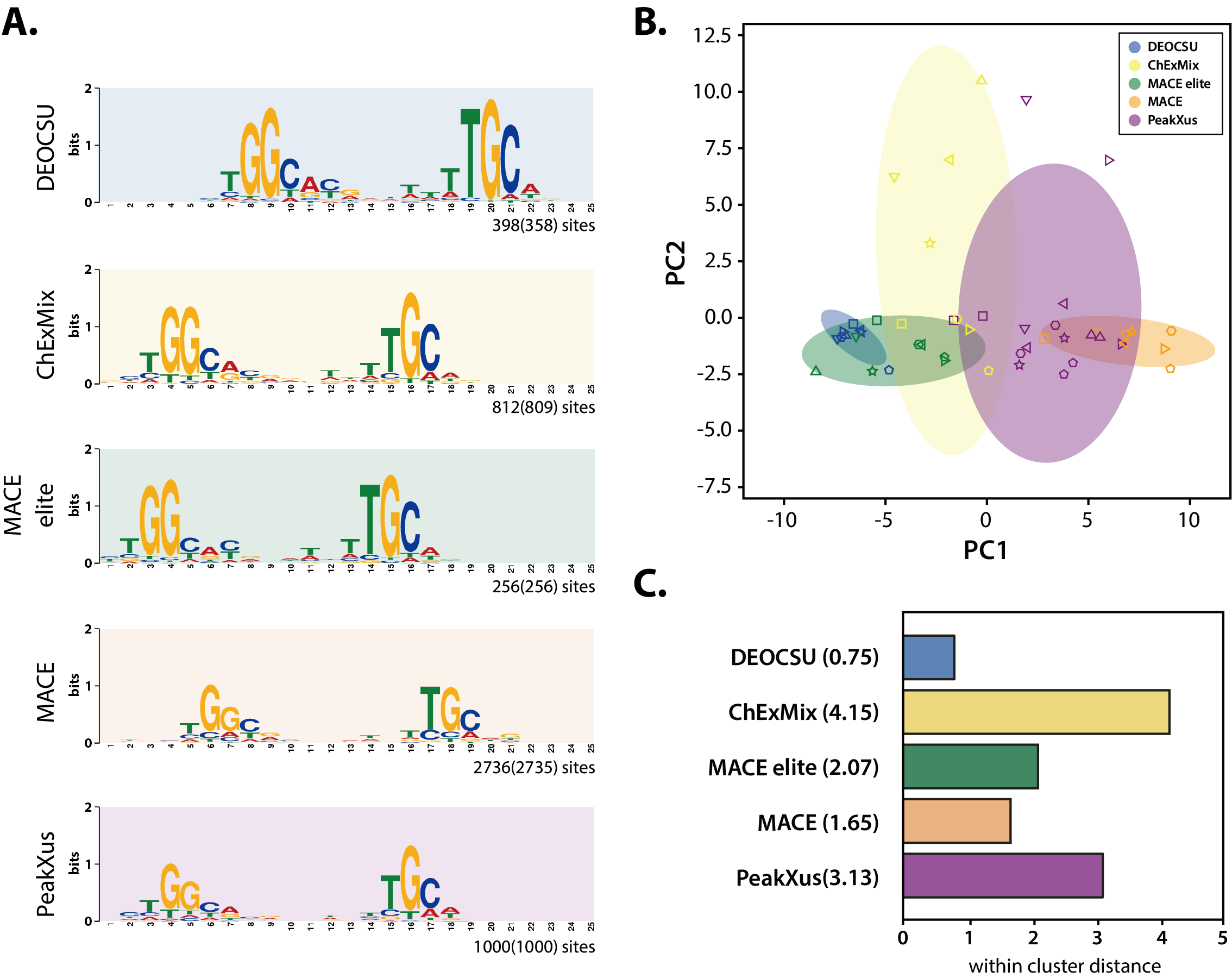
그림 3. RpoN ChIP-exo 데이터 세트를 사용한 DEOCSU와 경쟁 소프트웨어 간의 선별 모티프 비교.(A) DEOCSU 및 기존의 소프트웨어에서 선별한 대장균 K-12 W3110 균주의 RpoN 모티프 서열 (B) 다양한 대장균주 (BL21(DE3), CFT073, Crooks, O157:H7 EDL933, HS, K-12 W3110)에서 결합 모티프의 문자 확률 행렬 값을 이용한 2차원 주성분 분석(PCA) (C) 각 소프트웨어의 선별 데이터에 대한 모티프의 클러스터 거리 비교 다양한 균주들에서 진행된 시그마 인자 RpoN에 대한 ChIP-exo 데이터 분석 결과에서 DEOCSU는 각 데이터에 대한 정확한 모티프 서열을 찾아낼 수 있는 것을 확인할 수 있었고, 가장 일관된 모티프 시퀀스를 나타내었음을 확인함. |
|
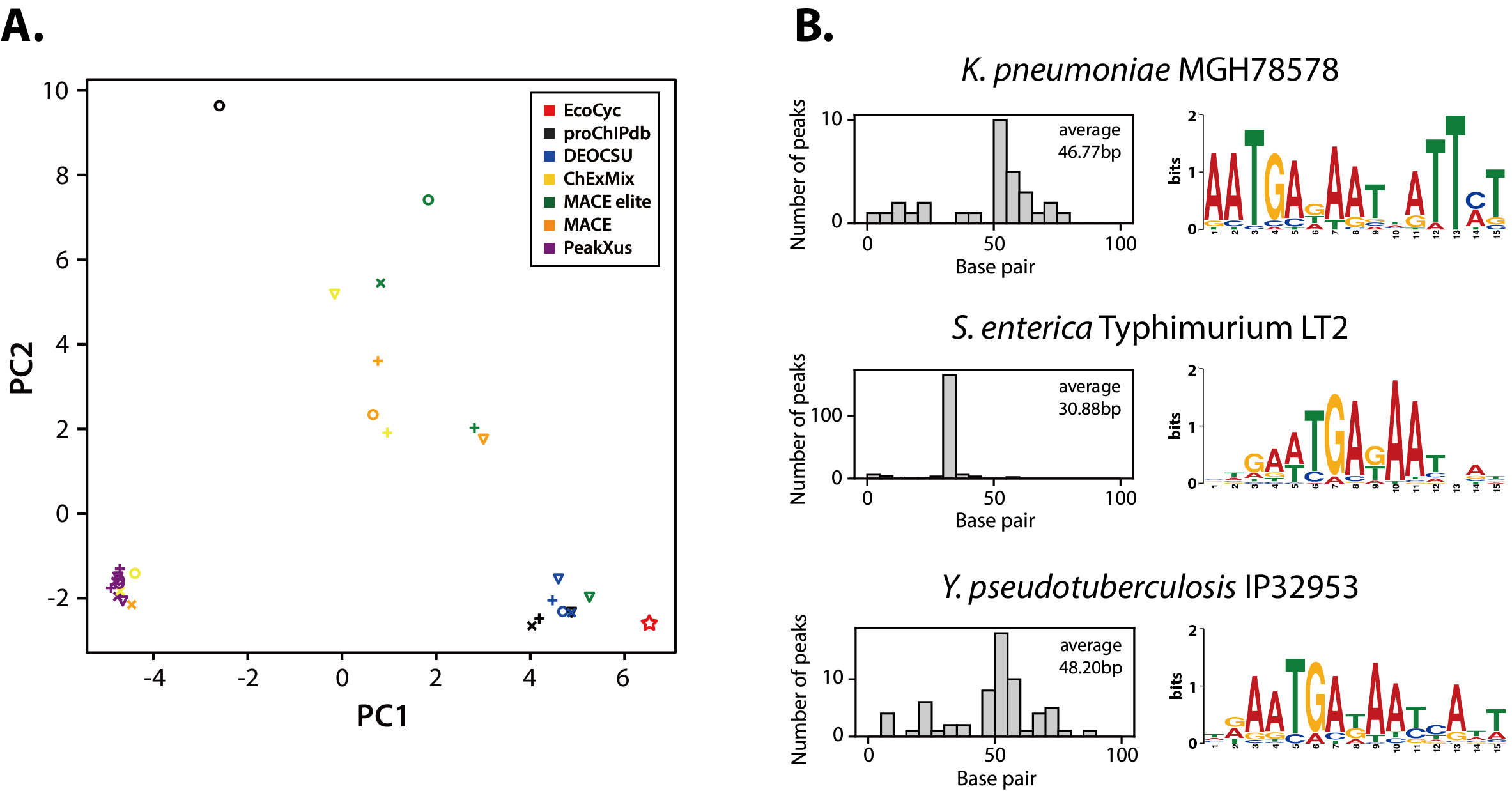
그림 4. 공개된 외부 원핵생물 ChIP-exo 데이터에 대한 DEOCSU의 분석 결과.(A) 네 가지 다른 탄소원 조건에서 Cra 결합 모티프의 문자 확률 행렬 값을 이용한 2차원 주성분 분석(PCA). DEOCSU의 학습데이터와 관계되지 않은 외부 전사 인자 ChIP-exo 데이터의 분석을 진행하였으며, 특히 Cra 데이터에서는 공개 데이터베이스(Ecocyc와 proChIPdb)의 결과와 가장 유사한 모티프 서열을 선별한 것을 확인할 수 있음. |
|
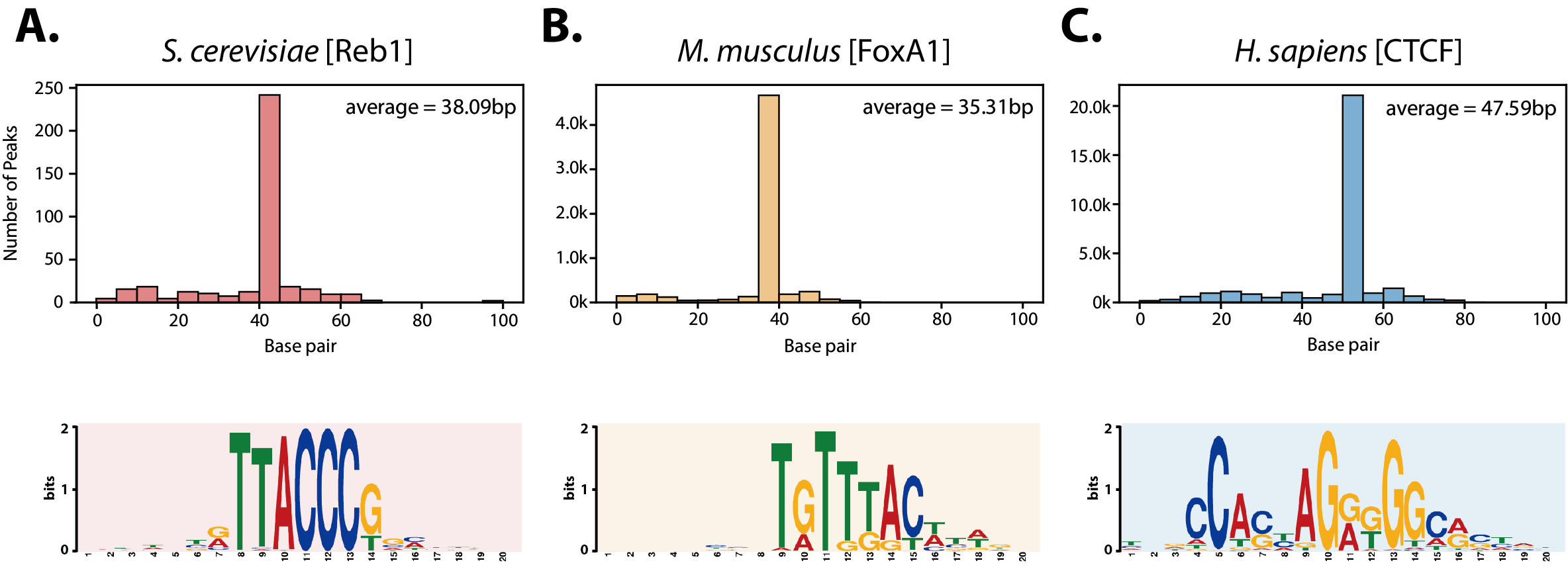
그림 5. 공개된 진핵생물 ChIP-exo 데이터에 대한 DEOCSU 분석 결과.(A) 효모(Saccharomyces cerevisiae Reb1), (B) 쥐(Musmusculus FoxA1) 및 (C) 인간(Homo sapiens CTCF)의 ChIP-exo 데이터에 대한 결합 폭 분포(위) 및 해당 모티브(아래). DEOCSU의 확장성을 확인하기 위해 진핵생물 데이터에 대한 분석이 추가적으로 진행되었으며, 해당 데이터 관련 논문에서 수동 선별을 통해 제안한 결합 폭 분포와 모티프를 유사하게 선별하는 것을 확인함 |
|
|
UNIST 홍보팀 news@unist.ac.kr TEL : 052)217-1230FAX : 052)217-1229 |




![[연구진] 김동혁 교수](https://news.unist.ac.kr/kor/wp-content/uploads/2014/11/press_20141125_04.jpg)
![[연구진] 이번 연구를 진행한 연구진의 모습. 왼쪽부터 이상목 박사후연구원, 방인아 연구원, 박서정 연구원](https://news.unist.ac.kr/kor/wp-content/uploads/2014/11/press_20141125_03.jpg)
![[연구그림1] DEOCSU의 개요](https://news.unist.ac.kr/kor/wp-content/uploads/2014/11/press03_20141125.jpg)
![[연구그림2] 대장균 K-12 MG1655 시그마 인자의 ChIP-exo 데이터를 이용한 DEOCSU의 성능 검증](https://news.unist.ac.kr/kor/wp-content/uploads/2023/02/연구그림2-대장균-K-12-MG1655-시그마-인자의-ChIP-exo-데이터를-이용한-DEOCSU의-성능-검증-151x55.png)