|
|
|
|
|
UNIST(총장 이용훈) 생명과학과 남덕우 교수팀은 단일세포 시퀀싱 데이터의 통합분석을 통해 폐암, 감염병 등의 질병과 관련된 유전자들을 효과적으로 선별해낼 수 있음을 밝혔다. 단일세포 시퀀싱(scRNA-seq)은 통상적인 벌크샘플 시퀀싱(RNA-seq)과는 달리 개별 세포수준에서 유전자 발현을 분석할 수 있는 기술이다. 특히 각종 질병, 발생, 분화 등 생물학적 과정을 분석하는데 널리 활용되고 있다. 벌크샘플은 상피세포와 각종 면역세포들이 섞여있어서 세포 유형의 차이를 고려하지 못하는 단점이 있다. 반면 단일세포 시퀀싱은 세포 유형별로 순일한 유전자 발현의 변화를 측정할 수 있어 질병의 발생 기작을 보다 정확하게 분석할 수 있다. 그러나 단일세포 데이터의 높은 잡음과 결측률, 데이터 간의 측정값의 차이(배치효과)로 인해 실제로 질병유전자의 분석에 얼마나 효과적인지 확인되지 않았다. 연구팀은 다양한 시뮬레이션 실험과 단일세포 데이터 분석을 통해 46가지 통합분석 방법을 비교했다. 특히, 국내에서 생산한 1기 폐암환자들의 단일세포(상피세포) 데이터를 세포유형별로 통합분석했다. 이를 통해 기존에 폐암관련 유전자로 보고된 90여 개 유전자들이 통계적으로 높은 순위를 가짐을 확인할 수 있었다. 이는 아직 밝혀지지 않은 폐암유전자들 또한 높은 순위로 선별할 수 있음을 시사한다. 이러한 결과는 기존에 진행됐던 수백 명의 벌크샘플 폐암환자의 데이터 분석에서도 달성하지 못한 결과로서 단일세포 통합분석이 폐암유전자 발굴에 매우 효과적이라는 것을 보여주고 있다. 연구팀은 이번 실험의 신빙성을 높이기 위해 대규모의 COVID-19 환자 샘플에서 단핵구 세포 10만개의 데이터를 추가로 분석했다. 통합분석으로 유전자 발현 데이터를 분석한 결과, COVID-19 바이러스의 침입에 대응하는 것으로 알려진 130여 개의 유전자가 통계적으로 높은 순위를 차지하는 것을 재차 확인할 수 있었다. 연구팀은 추가 실험을 통해 서로 다른 유형의 질병에서 단일세포 데이터의 통합분석이 질병유전자를 효과적으로 선별해 줄 수 있다는 것을 확인했다. 남덕우 생명과학과 교수는 “이번 연구 결과는 공개된 단일세포 데이터의 대규모 통합분석을 통해 새로운 질병 유전자들의 작용 경로를 발굴할 수 있음을 제시해주고 있다”며 “암, COVID-19 등 다양한 질병의 기작 연구에 단일세포 데이터를 세포 수준을 넘어 유전자 수준에서 더 적극적으로 활용할 필요가 있습니다”라고 밝혔다. 이번 연구는 UNIST 생명과학과의 Hai Nguyen 박사와 백부경 박사가 공동 1저자로서 수행했고 중견연구자 사업과 포스트게놈 다부처유전체사업에서 지원받았다. 연구결과는 세계적인 학술지 Nature Communications에 2023년 3월 21일자로 게재됐다. (논문명: Benchmarking integration of single-cell differential expression) |
|
|
|
[붙임] 연구결과 개요 |
1. 연구배경단일세포 시퀀싱(scRNA-seq) 기술은 벌크샘플 시퀀싱(RNA-seq)과 달리 개별 세포별로 시퀀싱을 수행하여 보다 높은 해상도로 유전자 발현을 측정하는 기술이다. 예를 들어 일반적인 암 샘플에는 암에 해당하는 상피세포와 T 세포, B 세포 등 각종 면역세포, 혈관세포 등이 섞여 있어서 RNA-seq 분석을 하면 여러 세포유형들의 평균적인 발현량을 측정해주는 반면, 단일세포 기술은 암 세포만 정확하게 골라서 유전자의 발현량을 측정할 수 있게 해준다. 따라서 단일세포 데이터 분석을 통해 암에서 유전자의 이상 발현을 보다 정확하게 분석할 수 있을 것으로 기대되고 있다. 그러나, 실제로 단일세포 데이터는 높은 잡음과 결측률을 나타내고 있고, 이러한 데이터의 분석이 벌크샘플 데이터 분석에 비해서 질병유전자들을 더 정확하게 선별해준다는 것을 체계적으로 입증한 연구는 없었다. 전 세계적으로 단일세포 데이터의 생산과 활용이 바이오-의료분야에 널리 확산되고 있다. 같은 질병에 관한 데이터라도 다른 연구실, 시간, 플랫폼 등에 따라서 측정값 사이에 기술적인 차이가 발생하는데 이를 배치효과라고 한다. 이러한 배치효과를 제거하고 서로 다른 단일세포 데이터들을 효과적으로 통합분석 하는 것이 중요한 연구과제가 되고 있다. 대부분의 단일세포 데이터 통합연구가 배치효과 제거 및 정확한 세포 분류에 집중되고 있고 유전자 수준의 통합분석 연구는 미미한 상황이다. 데이터 통합 후 결국 중요한 것은 세포유형별로 질병에 중요한 역할을 하는 유전자와 경로를 발굴하는 것이라서 유전자 수준의 효과적인 통합분석 연구가 매우 중요하다. |
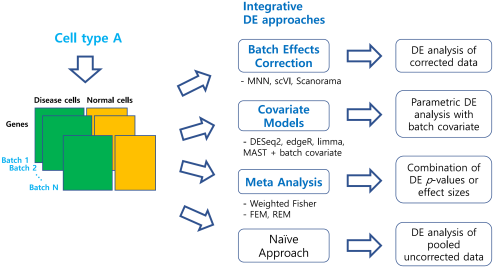
2. 연구내용이번 연구에서는 배치효과가 있는 여러 단일세포 데이터를 통합하여 주요 유전자를 발굴할 수 있는 46가지의 계산 파이프라인들의 주요 유전자 예측 정확도를 비교하는 연구를 수행하였다. 다음과 같은 세 가지 접근방법을 고려하였다: (1)데이터변환을 통한 배치효과 제거, (2) 통계모델에 배치공변인 포함, (3) 메타분석 (그림 1). 다양한 실험조건의 시뮬레이션 데이터를 생성하여 F 점수, precision-recall 분석, false-positive control, 계산시간 등 다양한 측면을 비교분석하고 실험 조건별로 최적의 계산방법들을 제시하였다. 특히, 한국인 폐암 단일세포 데이터를 통합분석하여 수백 명의 대규모의 벌크샘플 데이터 분석에 비해 훨씬 정확하게 폐암유전자들을 예측할 수 있음을 통계적으로 입증하였다. 또한, COVID-19 환자들의 단일세포 단핵구 데이터 10만개를 분석하여 바이러스에 대응하는 유전자 130여개를 통계적으로 유의한 높은 랭크로 선별할 수 있었다. |
3. 기대효과실용적인 관점에서 다양한 실험조건에서의 시뮬레이션 분석과 실제 데이터 분석을 통해, 최적의 단일세포 데이터 통합분석 알고리즘들을 제시하였고, 단일세포 데이터의 통합분석이 질병유전자 발굴에 통계적으로 유의한 예측력을 가진다는 것을 최초로 입증하였다. 단일세포 데이터의 많은 잡음과 결측치로 인해 대부분의 연구가 정확한 세포분류에 머물고 있지만, 이번 연구를 통해, 단일세포 데이터 통합을 통한 질병유전자 발굴에 토대를 제공해줄 수 있을 것으로 기대된다. |
|
[붙임] 용어설명 |
1. 단일세포 시퀀싱 (scRNA-seq)단일 세포 시퀀싱은 각각의 세포 수준에서 전체 유전자의 발현을 측정하는 기술입니다. 세포 단위에서 다르게 발현되는 유전자를 식별함으로써 조직과 기관 내 세포의 다양성을 파악할 수 있으며, 세포 수준에서 질병 상태를 조사할 수 있습니다. 단일 세포 RNA 시퀸싱은 기존의 벌크 샘플 시퀸싱과 비교하여 기술적으로 복잡하고 비용이 많이 들며 잡음과 결측치가 높아 분석의 난이도가 높다는 단점이 있습니다. |
2. RNA 시퀀싱 (RNA-seq)RNA 시퀀싱은 특정한 생물학적 조건에서 유전체가 생성하는 전체 RNA 전사체(transcriptome)를 분석하는 기술로 유전자 발현에 대한 정량적인 분석을 제공한다. 벌크 샘플 시퀸싱은 샘플 내에 포함된 모든 세포의 평균적인 유전자 발현을 측정하는 기술로 단일세포 시퀸싱과 비교했을 때 보다 단순하고 비용이 적게 들지만 샘플 내에 포함된 여러 세포유형들의 차이를 고려하지 못한다는 단점이 있다. RNA 시퀸싱은 유전자 발현 분석을 통해 질병과 약물 등 다양한 생물학적 조건에서 타겟이 될 수 있는 바이오 마커를 발굴하거나, 발현하는 유전자 양상을 통해 질병을 진단하는 등 생물학 및 의학 분야에서 폭넓게 사용되고 있다. |
3. 배치효과 (batch effects)배치효과란 시퀸싱 데이터 내의 여러 샘플 그룹(배치) 사이에 나타나는 기술적인 차이를 말합니다. 이러한 차이는 시퀀싱 기기, 실험 프로토콜, RNA 추출 방법, 온도 또는 습도와 같은 실험실 환경의 차이를 포함한 다양한 요인으로 인해 발생할 수 있습니다. 배치효과가 존재하는 RNA 시퀸싱 데이터를 분석할 경우 유전자가 세포 및 샘플 간 다르게 발현하는 양상이 배치효과에 영향을 받을 수 있어 잘못된 결과가 도출될 수 있다. 데이터의 기술적 변이를 줄이고 분석의 신뢰성을 높일 수 있도록 배치 효과를 해결할 수 있는 배치정규화 기법이나 배치 보정 방법 등 다양한 방법이 개발되고 있다. |
|
[붙임] 그림설명 |
|
그림1. 단일세포 데이터 통합분석 파이프라인 비교. 배치보정, 공변인 모델, 메타분석 등 46가지 계산 파이프라인의 주요유전자 예측 정확도 비교 |
|
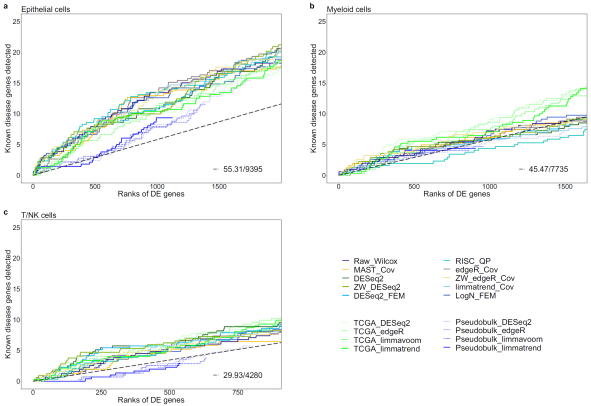
그림2. 단일세포 데이터 통합분석을 통한 폐암유전자 예측 정확도 비교. 초록색선들이 수백명의 대규모 벌크샘플을 이용한 예측으로서 단일세포 데이터 통합분석보다 예측정확도가 떨어지는 것을 알 수 있다 (Epithelial). Myeloid와 T/NK 세포 데이터에서는 폐암유전자에 대한 예측력이 거의 없다는 것을 보여준다. |
|
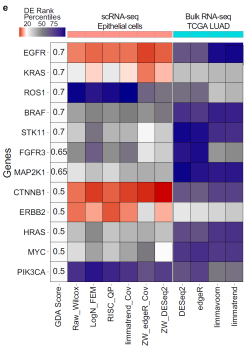
그림3. 단일세포와 벌크샘플 데이터 분석을 통한 주요 폐암유전자들 랭크 비교. |
|
그림4. 10만개의 COVID-19 단핵구 데이터를 이용한 바이러스 반응 유전자 예측 정확도 비교. 대부분의 단일세포 분석 방법들이 통계적으로 유의한 질병유전자 예측력을 보여준다. |
|
|
UNIST 홍보팀 news@unist.ac.kr TEL : 052)217-1230FAX : 052)217-1229 |





![[연구그림1] 단일세포 데이터 통합분석 파이프라인 비교](https://news.unist.ac.kr/kor/wp-content/uploads/2014/11/press_20141125_03.jpg)
![[연구그림2] 단일세포 데이터 통합분석을 통한 폐암유전자 예측 정확도 비교](https://news.unist.ac.kr/kor/wp-content/uploads/2014/11/press03_20141125.jpg)
![[연구그림3] 단일세포와 벌크샘플 데이터 분석을 통한 주요 폐암유전자들 랭크 비교](https://news.unist.ac.kr/kor/wp-content/uploads/2023/04/연구그림3-단일세포와-벌크샘플-데이터-분석을-통한-주요-폐암유전자들-랭크-비교-151x214.png)