
“지금의 통계기법으로 찾아낸 질병 유전자 후보군은 ‘빙산의 일각’에 불과합니다. 물속에 잠긴 큰 빙산까지 발굴하기 위해 효과적인 통계 알고리즘을 개발했죠.”
남덕우 생명과학부 교수팀은 최근 질병 유전자 후보군을 ‘정확하게 많이’ 찾아내는 통계 알고리즘(GSA-SNP2)을 개발했다. 이 알고리즘은 만 명 이하의 적은 유전체 데이터만 있어도 효과적으로 작동한다. 질병 유전자 후보군을 발굴하는 비용과 시간을 크게 줄일 방법으로 주목받고 있다.
남덕우 교수는 “몇 천 명 단위에서도 의미 있는 유전자 그룹을 찾아낼 수 있는 저비용 고효율 통계분석도구”라며 “이 알고리즘을 통해 신약 개발을 위한 유전자 표적을 발굴하거나 질병에 대한 이해를 더 빠르게 진행시킬 수 있다”고 강조했다.

사람의 DNA 염기서열의 차이를 가져와 통계적으로 분석하면, 특정 질병 등에 관여하는 유전자를 찾아낼 수 있다. 이런 작업에는 효율적인 통계 알고리즘이 꼭 필요하다. 사진은 남덕우 교수팀이 GSA-SNP2를 두고 상의하는 모습이다. | 사진: 김경채
사람마다 DNA 염기서열은 조금씩 다르게 나타나는데, 이로 인해 질병에 대한 감수성 등 다양한 표현형이 결정된다. 이런 염기서열의 차이를 ‘스닙(SNP)’이라고 하며, 대규모 유전체 데이터를 통계적으로 분석하면 특정 질병과 관련된 스닙(SNP)들을 찾을 수 있다.
천문학적인 비용과 시간을 들여서 데이터를 생산해도, 현재 사용하는 통계분석 방법들은 유의미한 스닙(SNP)을 많이 찾지 못한다. 질병 유전자가 아닌데, 질병 유전자로 판단하는 ‘허위양성’ 결과를 엄격하게 통제하도록 설계됐기 때문이다. 결국 수만 명의 유전형 데이터를 생산하고, 수십만에서 백만 개 이상의 스닙(SNP)을 대상으로 분석해도, 질병 유전자 후보군 수십 개 정도를 얻는 데 그치게 된다.
남 교수는 “허위양성을 통제해서 정확한 결과를 얻는 것도 중요하지만, 너무 많은 스닙(SNP)을 걸러내면 실제 신약개발 등에서 효용성이 낮아진다”며 “질병 유전자 후보군을 많이 발굴해낼 수 있는 ‘통계적 예측력(statistical power)’도 높여야 실용적인 통계 알고리즘이 된다”고 말했다.
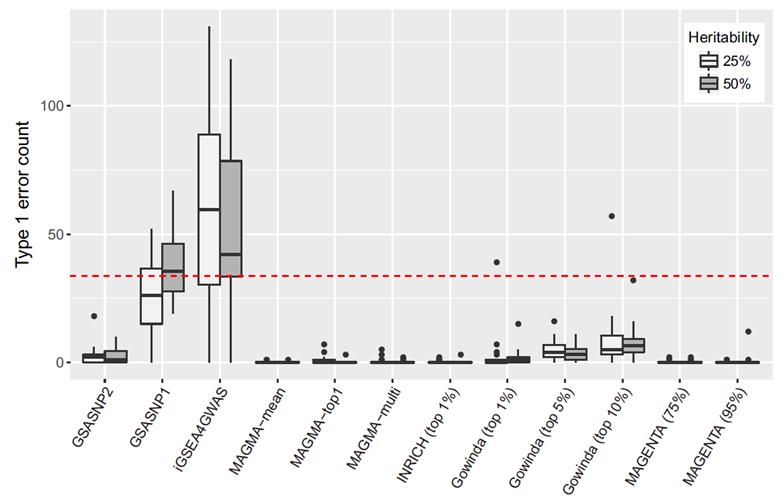
남 교수팀에서 개발한 통계 알고리즘 GSA-SNP2는 기존 마그마나 마젠타 프로그램만큼 정확도가 높다.
남 교수팀은 허위양성을 잘 통제해 정확한 결과를 얻으면서도 통계적 예측력을 높이는 알고리즘 개발을 목표로 삼았다. 이를 위해 ‘유전자 그룹(pathway) 상관관계 분석법’을 활용하면서 유전자 스코어에 큐빅 스플라인(cubic spline) 이라는 수학적 보정을 적용했다.
유전자 그룹은 특정 기능을 수행하는 데 관여하는 유전자 집단이다. 이들은 수백에서 수천가지 그룹들로 선별돼 데이터베이스로 정리돼 있다. 이 정보를 이용하면 개별 스닙(SNP) 비교에서는 놓쳤던 의미를 새롭게 찾을 수 있다. 남 교수팀은 이 기법을 쓰면서, 이미 질병과 상관관계가 높게 나타난 스닙(SNP)들은 제외하고 유전자 스코어를 보정함으로써 통계적 예측력을 높였다.
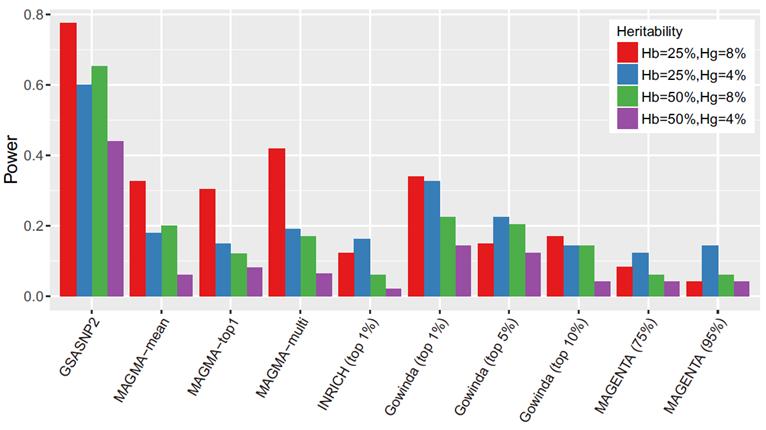
GSA-SNP2는 기존 통계 알고리즘보다 훨씬 많은 유전자 후보군을 골라낼 수 있어, 신약 표적 등을 설정하는 연구에 효과적으로 적용될 수 있다.
남 교수는 “질병 유전자 후보로 강하게 판단되는 스닙(SNP)을 빼면, 임의의 유전자 분포(영 분포)를 얻게 된다”며 “이 상태에서 다시 통계적으로 유의미한 걸 찾아내도록 설계했기 때문에 기존 방법들보다 2배 이상 예측력이 높아졌다”고 설명했다.
그는 이어 “새로운 통계 알고리즘을 적용하면 다양한 질병 유전자 그룹들을 다수 발굴할 수 있을 것”이라며 “신약개발 또는 스닙(SNP)분석 관련 연구기관이나 기업에서 활용하면 질병 치료에 기여하는 유용한 도구가 될 것”이라고 덧붙였다.
이번 연구는 UNIST 생명과학부의 윤소라 대학원생과 응우옌 하이(Nguyen C. T. Hai) 박사가 공동 1저자로 수행했으며, 포스트게놈 다부처유전체사업에서 연구비를 지원 받았다. 연구결과는 영국 옥스퍼드대학 출판사에서 발행하는 저명한 생물학 저널 ‘뉴클레익 에시드 리서치(Nucleic Acids Research, IF: 10.162)’ 3월 19일자 온라인판에 게재됐다.
















{kind=link}