
A research team, affiliated with UNIST has proposed a novel deep neural network (DNN) that can be successively applied to determine the saddle point without using conventional field-update algorithms. Their findings are now made freely available to the public. They are expected to contribute to the developments in polymer field-based simulations, according to the research team. This breakthrough has been led by Professor Jaeup U. Kim and his research team in the Department of Physics at UNIST.
In this work, the research team proposed a new deep learning (DL) approach that predicts the difference between the input and saddle-point pressure fields. According to the research team, their NN is designed to retain its predictive power at various incompressibility error levels so that it is less sensitive to the Langevin step interval. “[The new] approach completely replaces the conventional field-update algorithms with deep NN so that the saddle point search can be completed without conventional relaxation methods, such as AM,” said the research team. “Consequently, the performance is greatly enhanced because our new approach utilizes the predictive power of NN several times during each Langevin step.”
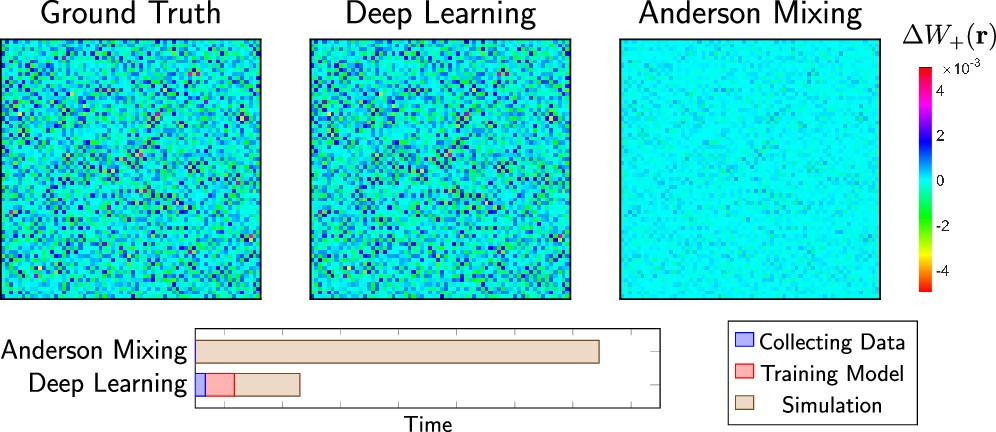
Figure 1. Comparison between Anderson Mixing (AM) and Deep Learning (DL). Above shows the two-dimensional slices of the ground truth ΔW+(r) and predictions of DL and AM, respectively, where the ground truth denotes an ideal output for the ML model.
The findings of this study have been published in the August 2022 issue of Macromolecules. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of the Ministry of Science and ICT (MSIT) and the Sejong Science Fellowship.
Journal Reference
Daeseong Yong and Jaeup U. Kim, “Accelerating Langevin Field-Theoretic Simulation of Polymers with Deep Learning,” Macromolecules, (2022).










{kind=link}