
A research team, led by Professor Dougu Nam in the Department of Biological Sciences at UNIST revealed that integrated analysis of single-cell sequencing (scRNA-seq) data can effectively select genes related to diseases, such as lung cancer and infectious diseases.
Single-cell sequencing (scRNA-seq) is a technology that can analyze gene expression at the individual cell level, unlike conventional bulk RNA sequencing (RNA-seq). In particular, it is widely used to analyze biological processes such as various diseases, outbreaks, and differentiation.
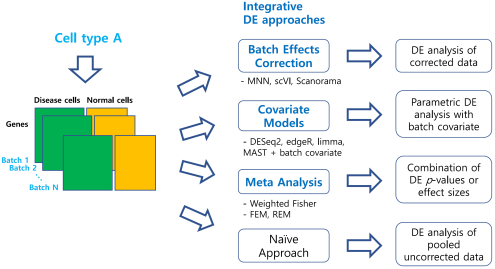
Figure 1: An overview of our benchmark study for differential expression (DE) analysis of scRNA-seq data with multiple batches. In total, 46 workflows from three integrative strategies and the naïve approach were tested.
Bulk samples have the disadvantage of not being able to consider the difference in cell type because epithelial cells and various immune cells are mixed. On the other hand, single-cell sequencing can measure changes in gene expression by cell type, which can more accurately analyze the mechanism of disease occurrence. However, due to the high noise and missing rate of single-cell data, and the difference in measurements (batch effect) between the data, it has not been confirmed how effective it is for the analysis of disease genes in practice.
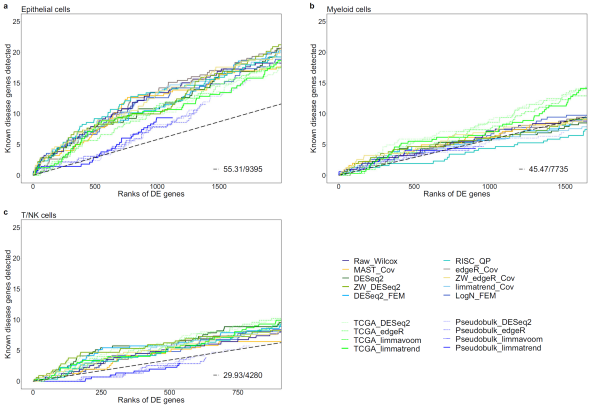
Figure 2. Comparison of predictive powers for lung adenocarcinoma (LUAD) genes between differential expression (DE) workflows for scRNA-seq and bulk RNA-seq data. Cumulative disease gene scores (GDA scores) for known disease genes up to top 20% DE gene ranks are shown for three cell types: a epithelial cells, b myeloid cells.
The research team compared 46 integrated analysis methods through various simulation experiments and single-cell data analysis. In particular, single cell (epithelial cell) data of domestic stage 1 lung cancer patients were integrated and analyzed by cell type. Through this, it was confirmed that more than 90 genes previously reported as lung cancer-related genes had statistically high rankings. This suggests that lung cancer genes that have not yet been identified can also be selected in a high order. These results were also not achieved in the previous data analysis of hundreds of bulk sample lung cancer patients, showing that single-cell integrated analysis is very effective in discovering lung cancer genes.
To increase the reliability of the experiment, the research team analyzed additional 100,000 mononuclear cells in a large sample of COVID-19 patients. As a result of analyzing gene expression data through integrated analysis, it was confirmed again that more than 130 genes known to respond to the invasion of the COVID-19 virus were statistically high. Through additional experiments, the research team confirmed that integrated analysis of single-cell data in different types of diseases can effectively select disease genes.
The findings of this research have been published in the March 2023 issue of Nature Communications. This work has been jointly participated by Hai C. T. Nguyen and Bukyung Baik in the Department of Biological Sciences at UNIST, as fist co-authors. It has been also supported by the National Research Foundation (NRF) of Korea and Genomics Programs.
Journal Reference
Hai C. T. Nguyen, Bukyung Baik, Sora Yoon, et al., “Benchmarking Integration of Single-cell Differential Expression,” Nat. Commun., (2023).










{kind=link}